

In the dynamic landscape of healthcare, embracing innovation remains paramount in our quest to tackle intricate medical issues effectively. One of the most promising frontiers of medical innovation is the development of combination products, which combine medical devices with biological components.
These innovative combination products have the power to reshape the landscape of medical diagnosis, treatment, and condition management, ushering in a new era of possibilities. However, their unique nature also presents a complex regulatory landscape that manufacturers and healthcare professionals must navigate.
What is a Combination Product?
Combination products represent a category of therapeutic and diagnostic items that integrate drugs, devices, and/or biological products. Within the realm of combination products, each component is designated as an integral constituent, playing a pivotal role in the overall functionality of the product
The primary purpose of combination products is to provide enhanced therapeutic benefits, improved patient outcomes, or more convenient administration compared to individual products.
There exist nine distinct categories of combination products, their classification hinging on their primary mode of action. Few are the following –
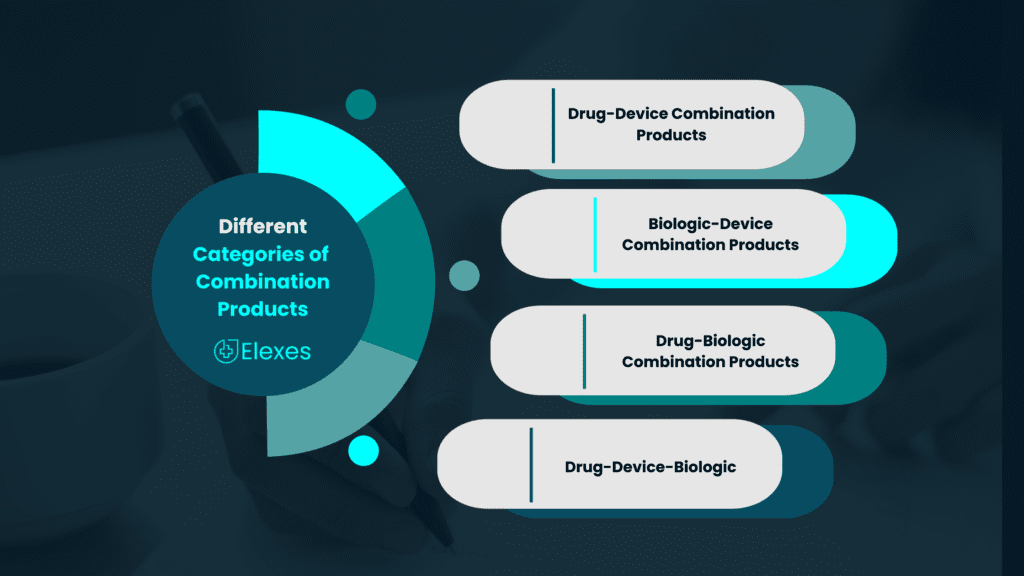
- Drug-Device Combination Products: These products combine a drug (pharmaceutical) component with a medical device. The device is often essential for the delivery, administration, or application of the drug.
Example: Insulin Pen - An insulin pen is a classic example of a drug-device combination product. It contains insulin (the drug) and a delivery device (the pen) that allows patients with diabetes to self-administer insulin subcutaneously. The device ensures precise dosing and convenient administration of the drug.
2. Biologic-Device Combination Products: These products combine a biological component, such as cells or tissues, with a medical device.
Example: Corneal Tissue Scaffold - A corneal tissue scaffold is a biologic-device combination product used in corneal transplantation. It incorporates donor corneal tissue (the biological component) and a specialized device that helps surgeons handle and position the tissue during surgery. This combination enhances the success of corneal transplants.
3. Drug-Biologic Combination Products: These products combine a drug and a biologic component, such as a monoclonal antibody, to enhance therapeutic efficacy.
Example: Anti-Tumor Monoclonal Antibody with Chemotherapy - In cancer treatment, some drug-biologic combination products combine a monoclonal antibody (biologic) with chemotherapy drugs (pharmaceutical). For instance, Rituximab, a monoclonal antibody, is combined with various chemotherapy agents to treat lymphomas. The antibody targets specific cancer cells while the chemotherapy drugs attack them systemically.
4. Drug-Device-Biologic Combination Products: These are complex products that incorporate a drug, a biological component, and a medical device, often used in advanced medical therapies.
Example: Artificial Pancreas System - An artificial pancreas system is a prime example of a drug-device-biologic combination product. It integrates continuous glucose monitoring (device) with insulin delivery (drug) and algorithms to regulate blood sugar levels. This system can include a biological component, such as glucagon, to counteract low blood sugar. It's used in the management of type 1 diabetes, mimicking the function of the pancreas.
What is not a Combination Product?
Several medical product types are commonly mistaken as “combination products” due to their complex nature, but they are not classified as such. Some examples include:
- Drug and Dietary Supplement Combinations: If a drug and a dietary supplement are used concurrently, they may appear as a combination product, but they are regulated differently, with drugs under the FDA’s purview and dietary supplements under a separate regulatory framework.
- Drug and Food: Some medical treatments involve specific dietary requirements while taking medications. While they work in tandem, they are subject to different regulations.
Understanding the Regulations for Combination Products
Due to ongoing technological advancements that blend these various product categories, the Food and Drug Administration (FDA) anticipates an increasing influx of combination products for evaluation.
This emerging phenomenon erases the conventional demarcations that have historically separated the FDA’s medical product centers, which notably include the Center for Biologics Evaluation and Research (CBER), the Center for Drug Evaluation and Research (CDER), and the Center for Devices and Radiological Health (CDRH).
The divergent regulatory pathways for each component can significantly influence every phase of product development and administration, including preclinical testing, clinical investigations, application for marketing approval, manufacturing and quality control, reporting of adverse events, promotional and advertising practices, and post-approval modifications.
What are the Typical Marketing Application Requirements for Combination Products in the US?
Typically, combination products gain marketing authorization by evaluating the primary mode of action (PMOA) exhibited by their individual constituent components.
💡 For example, when the primary mode of action stems from the drug component, the typical regulatory pathway involves the submission of a New Drug Application (NDA) or an Abbreviated New Drug Application (ANDA).If the biologic component holds the PMOA, a Biologic License Application (BLA) is necessary.
Alternatively, if the device component serves as the primary mode of action, it may require a premarket approval application (PMA), de novo certification, or premarket notification (often referred to as "510(k)".
Normally, a single marketing application suffices for a combination product. Nevertheless, there are instances where a sponsor might opt to submit separate marketing applications for distinct constituent parts, and the FDA may consider this approach permissible.
Steps for the US Market Entry
The initial stage involves the identification of the predominant mode of action exhibited by the combination product. This helps determine which FDA center will take the lead in the regulatory review:
Are you not clear or if both components of your combination product have significant roles? Contact us now, We stand ready to assist you in discerning the most fitting regulatory route for your product.
Request a Designation
If the manufacturer of combination products wants a definitive decision from the FDA concerning classification and/or assignment to a specific center, the sponsor can send a formal request for designation (RFD) to the Office of Combination Products (OCP). Alternatively, if the manufacturer seeks informal feedback, a pre-RFD can be filed to OCP.
We recommend initiating this request promptly, as soon as there exists adequate information for the FDA to render a judgment.
The request should include:
- Sponsor’s identity, including company details, contact person, and contact information.
- Product description, including classification, names, composition, developmental work results, manufacturing processes, proposed use, modes of action, and related products’ status.
- The sponsor’s suggestion regarding which FDA component should assume primary jurisdiction is rooted in the product’s most critical therapeutic action. If uncertain, follow the FDA’s assignment algorithm and assess other combination products for reference.
All submissions related to this should be addressed to the product jurisdiction officer and clearly marked as “Request for Designation.”
Once the FDA receives RFD, the following process occurs:
- Completeness Review: Within 5 working days of receiving the request, the FDA reviews it for completeness. In instances of incompleteness, the submission is returned to the sponsor along with a request for the requisite missing information.
- Designation Letter: Within a 60-day timeframe from the filing date, the FDA’s product jurisdiction officer issues a “Designation Letter” to the sponsor. This communication explicitly outlines the agency component entrusted with premarket review responsibilities, as well as any auxiliary consulting agency components involved in the process. If the FDA doesn’t issue this letter within 60 days, the sponsor’s recommendation for the primary jurisdiction center becomes the designation.
- Reconsideration Request: In case of a sponsor’s disagreement with the designated jurisdiction, they retain the option to request a reevaluation from the product jurisdiction officer. This appeal must be initiated within 15 days of the receipt of the designation letter. The request should not exceed 5 pages and cannot include new information. The FDA reviews and responds to this request within 15 days.
- Effect of Designation Letter: The designation letter is a firm determination, but it can be changed under certain circumstances. The FDA can change the designated agency component with the sponsor’s written consent or without it if it’s necessary for public health or compelling reasons. The sponsor is given notice and an opportunity to object, with specific timelines and the possibility of a meeting.
File the Application and FDA Approval/Clearance
Prepare a comprehensive application that includes data on safety, efficacy, manufacturing, labeling, and any other relevant information. The application should address the requirements specific to the chosen regulatory pathway (e.g., PMA, BLA). Submit the application to the FDA, paying attention to submission requirements and timelines.
Quick Tip - When it comes to RMAT combination products, if the device component is individually packaged, it might be appropriate to consider separate marketing applications for each of these constituent products. This is especially relevant when the delivery device may be used with multiple RMATs that share similar characteristics and administration requirements.
The FDA will conduct a thorough review of the application, involving experts from the relevant centers. Review timelines and requirements can vary based on the regulatory pathway and product complexity.
After review, the FDA will issue a decision on the approval, clearance, or rejection of the combination product. If approved, the product can be marketed in the United States.
Feedback Mechanisms Available
- Application-Based Mechanisms:
2. Combination Product Agreement Meetings (CPAMs):
The FDA discourages CPAM requests when there is an active FDA review of the combination product application, suggesting the use of application-based mechanisms at that stage.
Postmarketing Safety Reporting for Combination Products
The final rule on PMSR pertains to combination products subject to FDA premarket evaluation.
It applies to two types of entities:
The key components of the rule include:

- Reporting Requirements Based on Application Type: These obligations are applicable to both applicants seeking marketing authorization for combination products and those seeking authorization for their constituent parts. The specific reporting criteria vary depending on the type of application submitted.
- Reporting Criteria Based on Constituent Parts: These reporting requirements are exclusive to Combination Product Applicants and hinge upon the specific constituent parts included within the combination product.
- Information Sharing: Constituent Part Applicants must share certain postmarketing safety information with each other.
- Submission Process: Within the rule, you’ll find detailed instructions on how both combination product applicants and those dealing with constituent parts should approach the submission of PMSR information to the FDA.
- Streamlined Reporting: The rule provides options for combining specific reporting requirements into a single report.
- Records Retention: The rule provides explicit details on the types of records that combination product applicants and those dealing with constituent parts are obligated to retain and the prescribed duration for such record-keeping.
To whom does it apply?
The PMSR Final Rule on Combination Products encompasses two distinct applicant categories:
Applicant and Application:
Combination Product Applicant:
Examples of Combination Product Applicants include companies with approved applications for various types of combination products, such as drug-eluting stents, pre-metered dry powder inhalers with drug cartridges, vaccines in pre-filled syringes, or products combining a laser system and a specific drug for photoactivation.
Consider, for instance, a combination product comprising a laser system (device) and a light-activated drug, where:
- One entity possesses the approved PMA for the laser system's use within this combination.
- Simultaneously, a distinct entity possesses the approved NDA for the light-activated drug's utilization within this specific combination.
Conclusion
The combination products represent a promising frontier in healthcare innovation, offering new ways to diagnose, treat, and manage a wide range of medical conditions. However, their unique dual nature presents a complex regulatory landscape that manufacturers must navigate to bring these products to market.
Understanding the regulations for combination products, choosing the appropriate regulatory pathway, and meeting the stringent requirements for safety and effectiveness are essential steps in the development and approval of these innovative products.
As technology continues to advance and new therapeutic possibilities emerge, the regulatory framework for combination products will continue to evolve. Manufacturers, healthcare professionals, and regulatory agencies must work together to strike a balance between innovation and patient safety in this exciting field.
Email us now, we shall connect with you to see how we can assist you in your regulatory pathway.



















