

Access to De Novo Summaries | A treasure for manufacturers
The De Novo pathway by the US Food and Drug Administration (FDA), is a risk-based classification process to classify novel medical devices for which there are no legally marketed predicate devices. It is one of the least used pathways by medical device manufacturers when bringing a product to market because of the associated data requirements, required capital investment (User Fees for a De Novo application is $96,644, whereas for a 510(k) it is $10,953 and for a small business it is $24,161 for De Novo and $2,738 for 510(k)), and potential extensive clinical data.
However, in recent years, we are starting to see the popularity trending towards change.
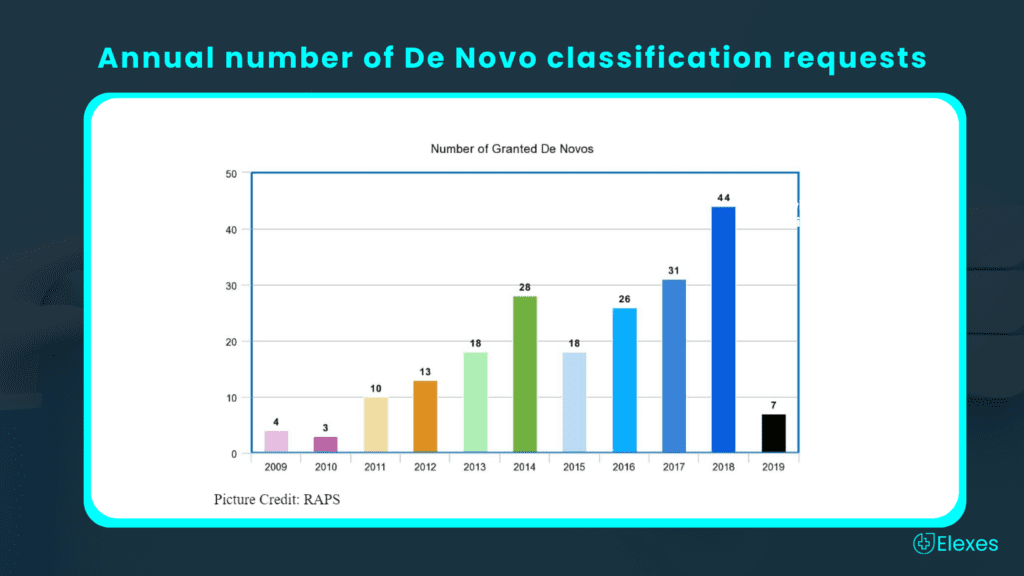
According to a RAPS report, trends show that the annual number of De Novo classification requests in recent years is steadily increasing and it hit a record of 44 De Novos granted during 2018.
As a part of easing the process of the De Novo pathway, the FDA published De Novo Classification Proposed Rule in December 2018, which seeks to provide structure, clarity, and transparency on the classification process. The FDA Commissioner Scott Gottlieb said, “Our goal is to make the De Novo pathway significantly more efficient and transparent by clarifying the requirements for submission and our processes for review.
As a result, we expect to see more developers take advantage of the De Novo pathway for novel devices.”. 235 safe and effective novel medical devices have been successfully marketed through the De Novo pathway since the FDA began granting marketing authorization for De Novo devices.
You must also check our blog on DeNovo paving the path for technological advancements to learn DeNovo classifications
The recent update from the FDA on April 23, 2019, Evaluation of Automatic Class III Designation (De Novo) Summaries, outlines the release of summary documents by the FDA for devices classified through the De Novo process. These summary documents are intended to have two significant purposes:
-
It serves as an objective and balanced summary of the scientific evidence that served as a basis for the decision to grant a De Novo request and
-
Serves as a resource for the manufacturers who may wish to use the device as a predicate for their future 510(k) submissions.
The De Novo summary documents in the FDA database includes the minimum of the following information:

These summaries serve as a reference database for the manufacturers planning for De Novo submissions. This provision of the FDA will provide manufacturers a better clarity in the De Novo classification process.
However, each of these summaries is only a part of the corresponding comprehensive De Novo submissions which need to be meticulously drafted and compiled to address all device-specific regulatory and statutory requirements.
Elexes has been assisting several medical device manufacturers in drafting, compilation, submission, and approval of De Novo. For further details please contact connect@elexes.com



















